Theme Cluster 1
Polyphilic molecules and their transitions in solutions
Competing Philicities: Cellulose solvation in polyphilic solvent mixtures (A1)
Supervisor: Prof. Daniel Sebastiani
Contact: daniel.sebastiani@chemie.uni-halle.de
The intriguing interplay of solvents with different philicities can be described empirically/ex-perimentally at a satisfactory level, while a comprehensive computational description still repre-sents a challenge. In principle, all necessary quantities are available, yet the accuracy in computing at the enthalpy level is not satisfactory, and there is no simple method known to describe the entropy of mixing of a simulation snapshot.
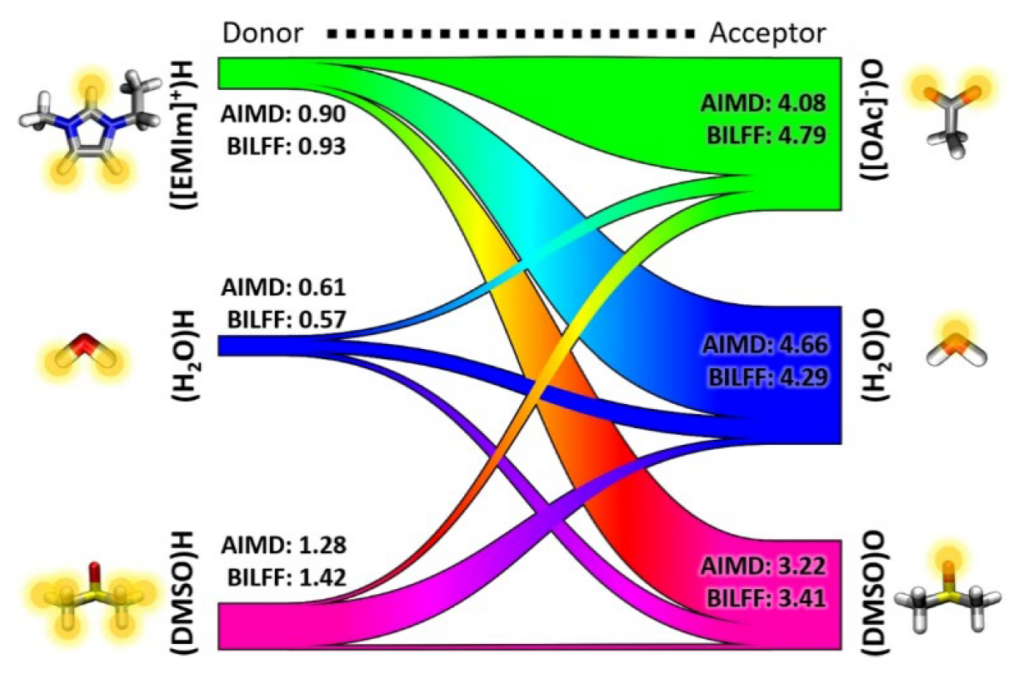
A particularly interesting system with a challenging combination of philicities is the process of cellulose solvation. Crystalline cellulose does not dissolve in common solvents (including water), despite its numerous hydrophilic groups. Current dissolution processes work with aggressive chemicals and under harsh conditions, with significant environmental drawbacks. A more environmentally friendly option for dissolving cellulose is given through ionic liquids and can be tuned by the addition of co- and anti-solvents. Cellulose can be considered a nearly limitless resource and dissolution is necessary for many industrial applications such as the production of biofuels and bioplastics.
The main objective of this project in the second funding period is the atomistic understanding of solvation processes of multi-component systems exhibiting multiple philicities, by means of molecular dynamics simulations. The more specific objective is to understand the solvation mechanism of cellulose in multi-component mixtures of ionic liquids, organic co-solvents, as well as anti-solvents such as water and ethanol.
Dynamic yet defined self-assembly of small molecules in solution: colloid-like ionic
clusters (ionoids) (A2)
Supervisor: Prof. Daniel Sebastiani & Prof. Dariush Hinderberger
Contact: daniel.sebastiani@chemie.uni-halle.de | dariush.hinderberger@chemie.uni-halle.de
We have recently discovered that small molecular building blocks with more than one interacting funtional group dynamically self assemble into non-trivial, non-random (thermal) structures in solution. This self-assembly into ionoids (as we termed them) takes place through a combination of preferential solvation, viscosity, van der Waals interactions together with long-range correlated electrostatics.
In this project in which two doctoral researchers will be supervised by Prof. Dr. Dariush Hinderberger and Prof. Dr. Daniel Sebastiani, we aim at deriving the full design principles that govern the formation of these dynamic structures from isotropic “clouds” of ions in solution through concepts of multiple non-covalent interactions.
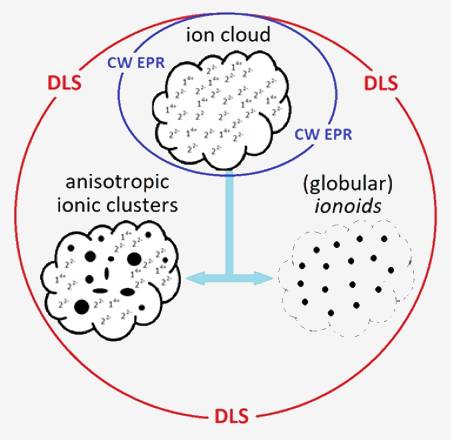
Experimental Physical-Chemical Project Main methods that will be used:
– Electron paramagnetic resonance (EPR) spectroscopy
– Dynamic light scattering (DLS) and Electrophoretic light scattering (ELS)
– Chemical synthesis of the ionoid building blocks
– Static light and potentially x-ray scattering (SLS, SAXS/WAXS)
– Other physico-chemical characterization methods (e.g. calorimetry)
– Mathematical (analytical) description of the ionoid-formation incubation process (e.g. based on bifurcation theory)
Going beyond the self assembly of small and medium-sized ionic molecules in solution, this project has developed three additional branches that deal with
– solvation of amphiphilic and fluorinated molecules in solution, i.e. the formation of aqueous lower polarity solvation shells (ALPSS)
– the complex processes in the formation of hydorgels, based on proteins, peptides or synthetic polymers, including solvation, hydrogen-bonding and ionic interactions
– a holistic description of protein-ligand binding in transport proteins like albumin or fatty acid binding proteins (FABPs).
Theme Cluster 2
Polyphiles in high symmetry structures – from solution to solid state
Complex self-assembly of polyphiles – Soft quasicrystals based on networks and honeycomb structures (A3)
Supervisors: Prof. Carsten Tschierske (experimental part) & Prof. Rebecca Waldecker (theoretical part)
Contact: carsten.tschierske@chemie.uni-halle.de & rebecca.waldecker@mathematik.uni-halle.de
Quasicrystals (QC) represent a state of condensed matter having long range order, but no translational periodicity. In previous work we have discovered a new type of liquid quasicrystals (LQC) combining triangular, square and trapezoidal cells. As well, first indications of LQC formation by combinations of only triangular and square cells with dodecagonal motifs were found. In the framework of this project we will search for new series of compounds being capable of forming LQCs by spontaneous self-assembly in the liquid state and to find new types of such self-assembled quasiperiodic patterns.
This will be achieved by a close collaboration between two PhD students, one focussing on theoretical and the other on experimental work.
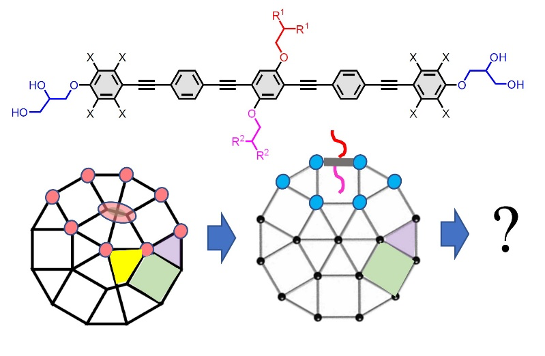
A relatively simple model for the OPE core case, capturing its geometric properties mostly, already gives us high predictive power for the synthesis. We intend to improve the model, adapt it to more cases, study the boundary cases for the parameters more closely and extend the modelling approach in two directions: integrating more details beyond geometry and including other types of molecules. In addition, we contemplate including simulations., which could lead to a
a working cycle like “choose or create suitable model – calculate model parameters – design – simulate – perform targeted synthesis – analyze”. Computer simulations based on MD simulations might deliver complementary views that will also allow a benchmark and competitive test for our models. To answer these questions, we need the design and (organic) synthesis of new material as well as detailed investigations of their molecular self-assembly, for example, by polarizing optical microscopy and X-ray scattering as well as by mathematical analysis of the structures, leading to new compounds, superstructures and improved models with higher predictive power.
Advancing the Cooperative Assembly of Amphiphilic Linkers: Probing Solvent Induced Ordering in Covalent Organic Frameworks (A6)
Supervisor: Jun.-Prof. Frederik Haase
Contact: frederik.haase@chemie.uni-halle.de
Covalent Organic Frameworks (COFs) are a promising class of materials with precise, crystalline structures that make them useful for applications such as catalysis, gas storage, and molecular separation.

However, the processes by which COFs form are still not fully understood, particularly the role solvents play during their assembly. This project explores how specially designed building blocks—amphiphilic linkers, which contain both hydrophilic and hydrophobic components—can guide the formation of COFs. By studying how these linkers interact with different solvent environments, we aim to uncover fundamental principles of how order emerges in these complex materials. This knowledge will not only advance the design of COFs with improved performance but also deepen our understanding of molecular self-assembly, a process central to both chemistry and biology.
Structure formation and pore-space properties of COFs with amphiphilic linkers (N1)
Supervisor: Prof. Kay Saalwächter
Contact: kay.saalwaechter@physik.uni-halle.de
Metal organic and covalent organic frameworks (MOFs and COFs, respectively) are intensely discussed as tailor-made functional materials for molecular storage/release, transport and separation, to be used in diverse fields such as (electro)catalysis, energy storage, drug delivery or (bio)sensing.Project A6 of Haase adopts an innovative strategy to create COFs with channels having different philicities by harnessing the self-organization of various types of pendant side chains of the linker molecules.

To explore the structure-directing effects and the application potential of such novel materials, this project proposes the application of a combination of solid-state NMR techniques to aid in structure elucidation and to provide a molecule-scale picture of the channel properties. On the one hand, the structure formation will be monitored via heteronuclear correlation techniques and methods characterizing the molecular dynamics of as well as polarization transfer between the building blocks (see figure). On the other hand, the channel properties are characterized by probing the partitioning and the dynamics of small molecules (e.g. solvent) with variable philicity.
Theme Cluster 3
Polyphilic molecules for enzymatic and chemical catalysis
Leveraging computational tools for non-natural enzymatic amination of polyphilic substrates (A5)
Supervisor: Prof. Martin Weissenborn
Contact: martin.weissenborn@chemie.uni-halle.de
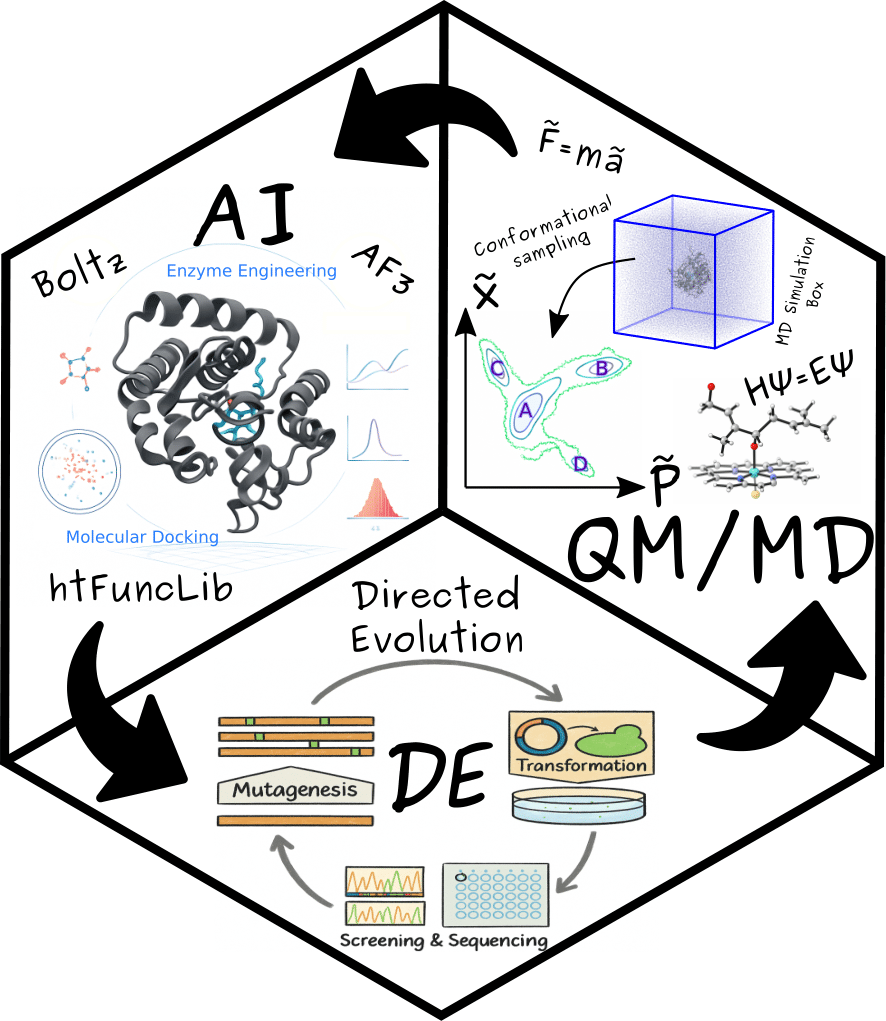
Understanding enzymes at the molecular level is essential for enhancing their catalytic efficiency and functional specificity. A high-fidelity approach integrates computational methods—such as molecular dynamics (MD) simulations, quantum mechanical (QM) calculations, and machine learning (ML) techniques—with experimental data, primarily from directed evolution. This synergistic framework offers unprecedented insights into the structural and dynamic properties of enzymes, enabling the rational engineering of catalysts with improved activity and selectivity.
Unspecific peroxygenases (UPOs) catalyze the oxyfunctionalization of a broad range of substrates by utilizing hydrogen peroxide (H₂O₂) as both an electron and oxygen donor. This unique capability makes them highly attractive biocatalysts for the selective transformation of complex organic molecules, including terpenes and steroids. However, their inherent substrate promiscuity underscores the need for precise engineering to fine-tune their chemical, regio-, and stereoselectivity for specific applications. This project leverages an integrated approach to identify critical determinants near the active site that govern substrate binding and selectivity, laying the groundwork for engineering UPO variants with enhanced specificity and catalytic performance.
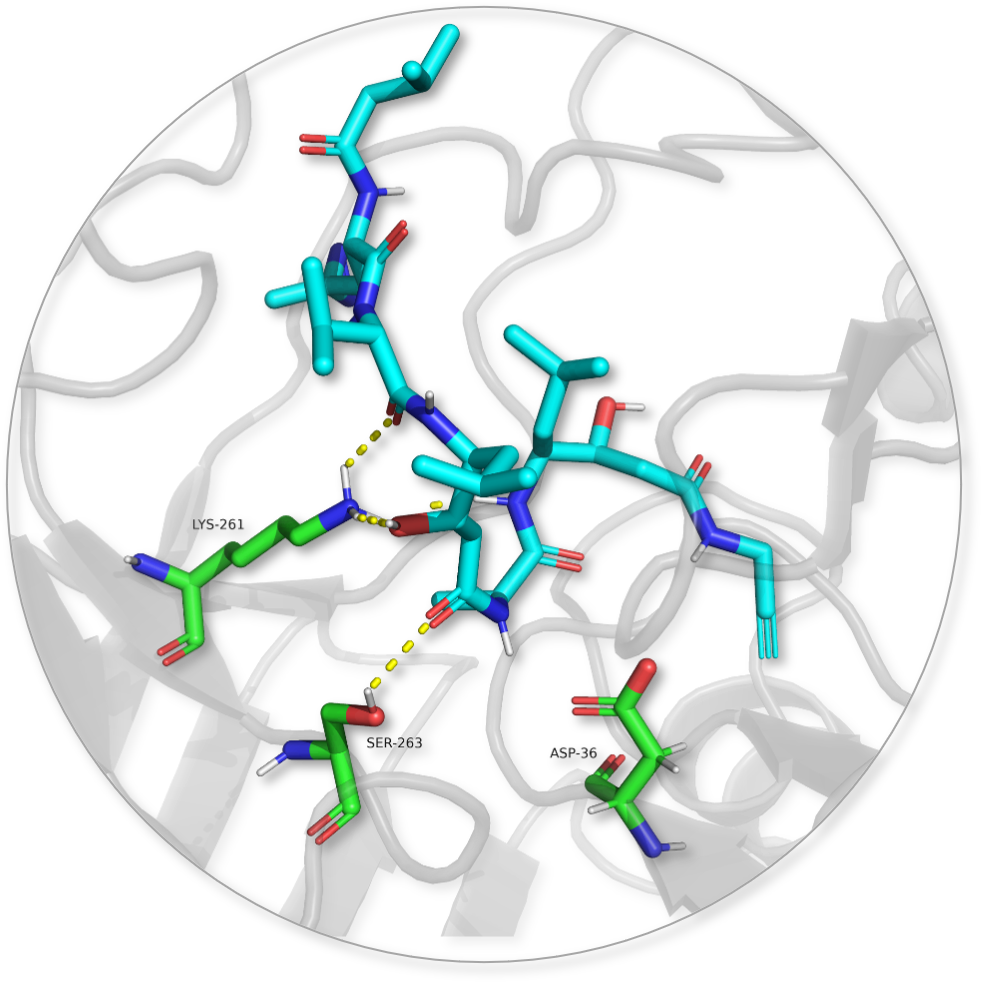
Dissecting the interactions that lead to selectivity of pepstatin-based probes for photoaffinity labelling of plant aspartic proteases (N2)
Supervisor: Dr. Mariana Schuster
Contact: mariana.schuster@ipb-halle.de
In this highly multidisciplinary project, we will study the polyphilic interactions that determine the specificity of pepstatin-based probes containing fluorinated amino acids for photoaffinity labelling towards plant aspartic proteases. To understand these determinants, we will harness the diversity of aspartic proteases from the model plant Arabidopsis thaliana and deploy chemical synthesis, advanced modelling, protease biochemistry, mass spectrometry and structural analysis methods. A detailed analysis of the consequences of this diversification will lead to an increased understanding of the polyphilic interactions in the active site of the proteases and would boost the development of better probes for aspartic proteases of plants and other organisms.
Active-Species-Switching in Micellar Catalysis via Control of Multiple Noncovalent Interactions (N3)
Supervisor: Prof. Robert Langer
Contact: robert.langer@chemie.uni-halle.de
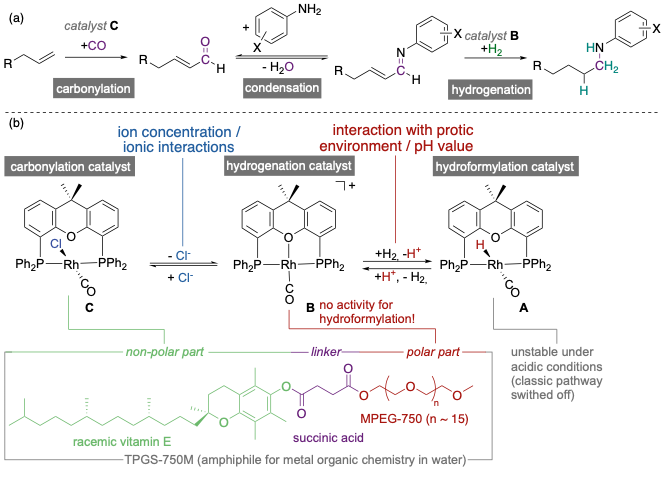
The major objective of this project is to provide a comprehensive picture of the parameters and interactions, leading to a switch of the operative mechanistic pathway in rhodium-catalysed hydroaminomethylation (HAM) reactions under micellar conditions. Within this project we will systematically investigate the influence of multiple non-covalent interactions on the catalytic performance of the rhodium-catalyzed HAM reaction in aqueous micelles, formed in the presence of different surfactants. By a combination of different experimental methods, we will determine the hydrodynamic radius, the formation of micelles or tubes, as well as the critical micelle concentration, depending on parameters critical for the observed catalytic reaction for different kinds of surfactants.
Theme Cluster 4
Polyphilic proteins and protein-like nanoparticles
Single-chain nanoparticles as novel catalytic entities (B2)
Supervisor: Prof. Wolfgang H. Binder
Contact: wolfgang.binder@chemie.uni-halle.de
Components in cells require a specific localized environment, so Nature has developed sophisticated entities on different scales to separate chemical reaction such as in enzymes, using eg. hydrophobic/hydrophilic pockets.
This project will design an opening/closing mechanism into an artificial polymer to mimic an artificial enzyme, wherein different philicities are arranged to enble a catalytic cycling inside a small nanoparticle (sized 10 nm).
- Synthesis of (resorcin[4]arene)6 to arrange the chemistry of resorcin[4]arene (aliphatic/fluorinated), and so adjust the philicities of the monomers to enable a proper hydrophobic encapsulation into the different compartments of the SCNPs. The exact location of the (resorcin[4]arene)6 cage in a specific compartment will be probed by STD NMR.
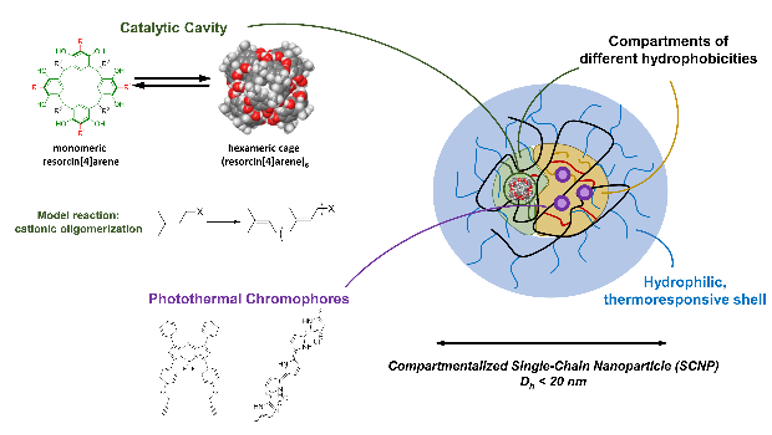
- Incorporation of (resorcin[4]arene)6-cages into SCNPs by (i) preformed cages, which are encapsulated into the SCNP-precursor-polymer prior to crosslinking the final SCNP; and (ii) using the formation of the (resorcin[4]arene)6 cages as crosslinker for the SCNP formation.
- Probing catalytic activity: The catalytic activity of the artificial enzymes will be studied on the catalytic cavities outside and inside the SCNPs. As model reaction the oligomerization of isoprenyl-diphosphate will be followed using HPLC-techniques with focus on chain length and kinetic parameters.
Dissecting Arf1 protein-protein and protein-membrane interactions (B4)
Supervisor: Prof. Kirsten Bacia
Contact: kirsten.bacia@chemie.uni-halle.de
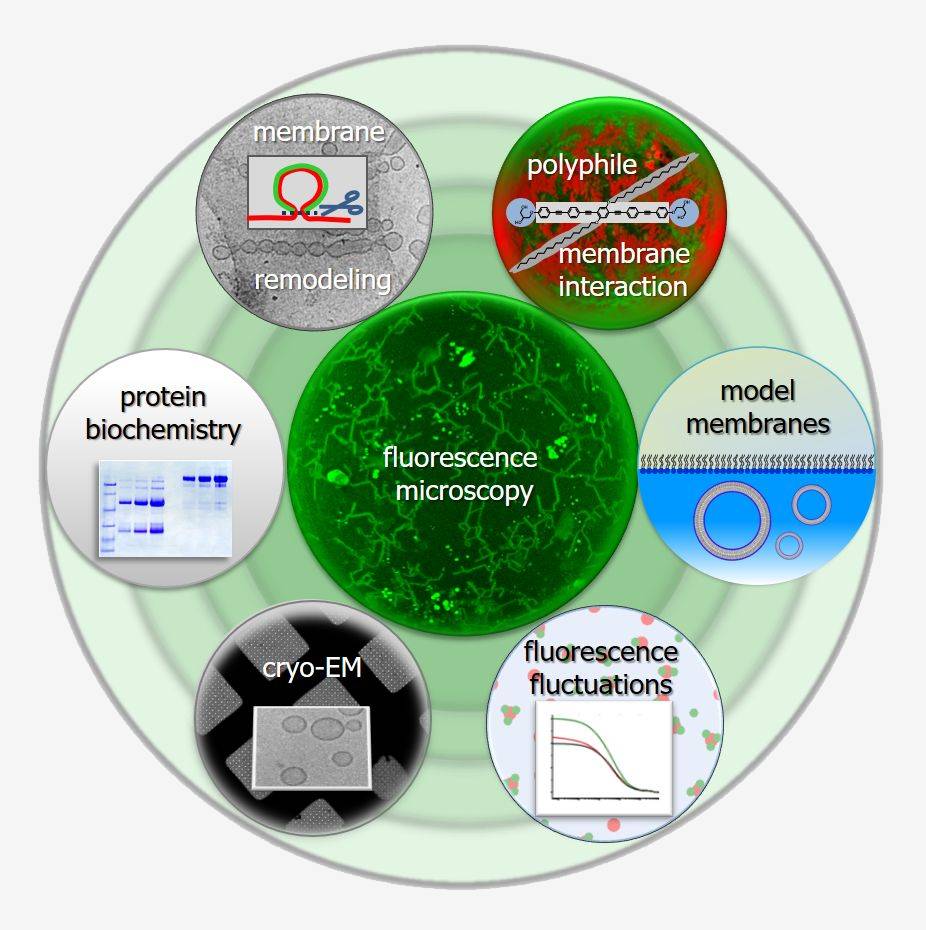
In biology, protein-protein interactions and protein-lipid interactions lead to highly complex, dynamically self-assembling and restructuring systems. The concept of amphiphilicity has been instrumental in gaining an understanding of biomembranes, including the fundamental self-assembly of the lipid bilayer and the integration of hydrophobic transmembrane domains of integral membrane proteins.
However, amphiphilicity alone does not suffice to describe membrane curvature generation by proteins that interact with bilayers through hydrophobic and amphipathic insertions and scaffolding. Further developing easily applicable concepts is therefore essential. In this project, we use the small GTPase Arf1, a molecular switch that features a membrane-interacting amphipathic helix and a myristoyl anchor and is able to form a curved scaffold, as a model protein. We investigate its polyphilicity as well as the interplay of multiple interactions between multiple Arf1 particles.